Fenyloketonuria Jest Warunkowana Autosomalnym Allelem Recesywnym A
Fenyloketonuria (PKU) to choroba metaboliczna uwarunkowana genetycznie, dziedziczona w sposób autosomalny recesywny. Oznacza to, że aby dziecko urodziło się z PKU, musi odziedziczyć wadliwy gen od obojga rodziców. Rodzice, którzy posiadają tylko jeden allel z mutacją genu, są nosicielami i zazwyczaj nie wykazują żadnych objawów choroby.
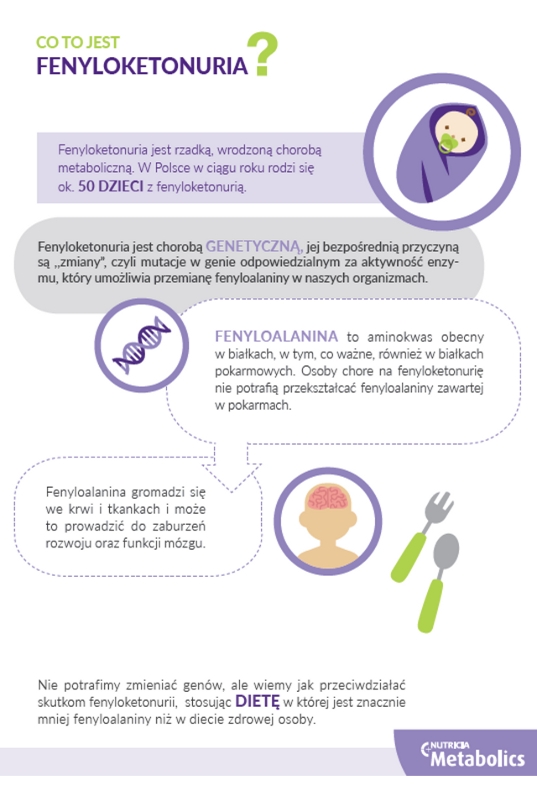
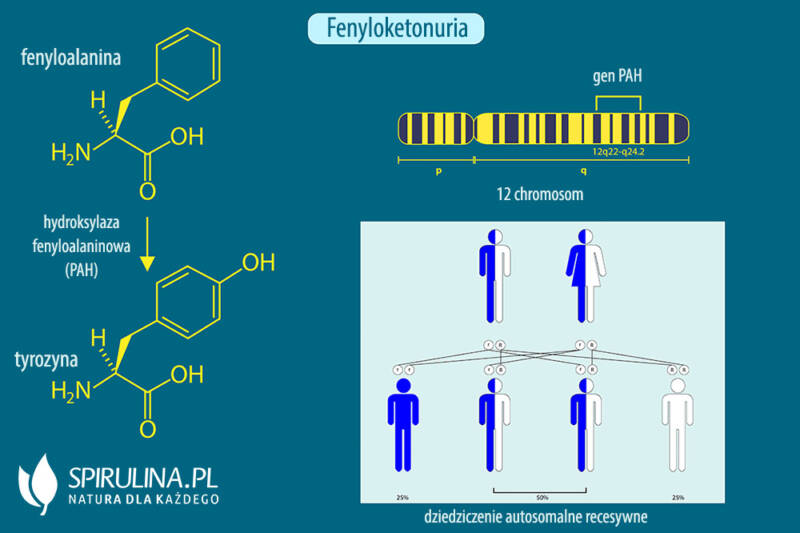
Fenyloketonuria spowodowana jest mutacją w genie PAH (hydroksylazy fenyloalaninowej), który koduje enzym odpowiedzialny za przekształcanie fenyloalaniny (aminokwasu zawartego w białkach) w tyrozynę. Brak lub niedobór tego enzymu prowadzi do gromadzenia się fenyloalaniny w organizmie, szczególnie we krwi i mózgu. Nadmiar fenyloalaniny jest toksyczny dla rozwijającego się mózgu, dlatego nieleczona PKU może prowadzić do poważnych uszkodzeń neurologicznych, takich jak opóźnienie rozwoju umysłowego, napady padaczkowe, zaburzenia zachowania i problemy z uczeniem się.
Dziedziczenie autosomalne recesywne – mechanizm i ryzyko
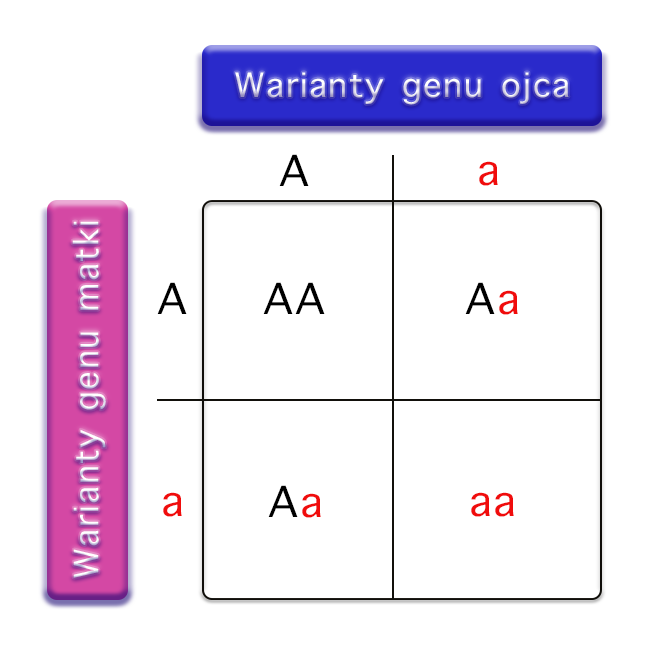
Zrozumienie mechanizmu dziedziczenia autosomalnego recesywnego jest kluczowe dla oceny ryzyka wystąpienia PKU u potomstwa. Każdy człowiek posiada dwie kopie każdego genu, jedną odziedziczoną po matce, a drugą po ojcu. W przypadku PKU, gen PAH znajduje się na chromosomie autosomalnym (czyli nie jest związany z chromosomami płci).
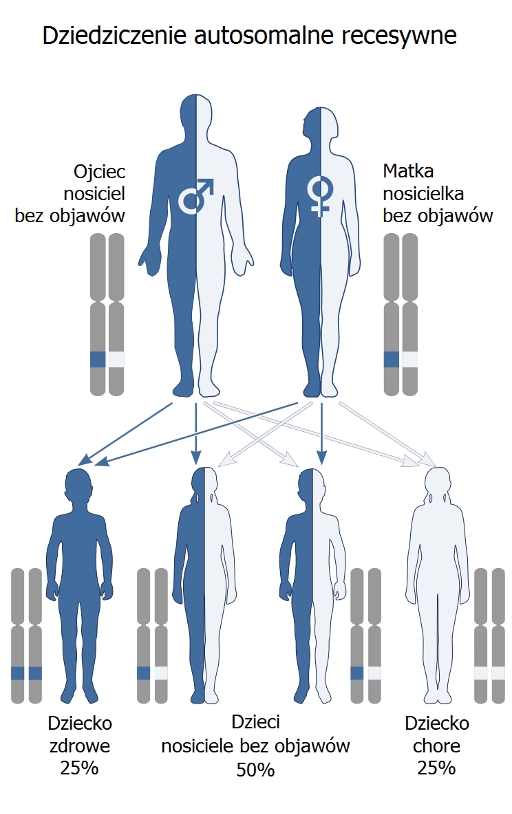
Osoba z dwoma prawidłowymi allelami genu PAH (PAH/PAH) jest zdrowa i posiada w pełni funkcjonalny enzym. Osoba z jednym prawidłowym allelem i jednym zmutowanym (PAH/pah) jest nosicielem. Posiada wystarczającą ilość funkcjonalnego enzymu, aby prawidłowo metabolizować fenyloalaninę, ale może przekazać zmutowany allel swojemu potomstwu. Osoba z dwoma zmutowanymi allelami (pah/pah) choruje na fenyloketonurię, ponieważ brakuje jej funkcjonalnego enzymu.
Jeśli oboje rodzice są nosicielami (PAH/pah), prawdopodobieństwo, że ich dziecko urodzi się z PKU (pah/pah) wynosi 25%. Istnieje 50% prawdopodobieństwo, że dziecko odziedziczy tylko jeden zmutowany allel (PAH/pah) i będzie nosicielem, a 25% prawdopodobieństwo, że odziedziczy dwa prawidłowe allele (PAH/PAH) i będzie zdrowe oraz nie będzie nosicielem. Ważne jest, aby pamiętać, że każda ciąża jest niezależnym zdarzeniem, a prawdopodobieństwo wystąpienia choroby w każdej ciąży pozostaje takie samo.
Przykładowo, jeśli oboje rodzice są nosicielami, a ich pierwsze dziecko ma PKU, prawdopodobieństwo, że kolejne dziecko również będzie chore, nadal wynosi 25%. Niektórzy ludzie mylnie zakładają, że po urodzeniu dziecka z PKU kolejne dzieci na pewno będą zdrowe. Jest to błędne przekonanie, wynikające z niezrozumienia probabilistycznego charakteru dziedziczenia.
Dlatego tak istotne jest przeprowadzanie badań przesiewowych noworodków. Badanie przesiewowe pozwala na wczesne wykrycie choroby jeszcze przed pojawieniem się objawów. W Polsce, jak i w wielu innych krajach, badanie w kierunku PKU jest obowiązkowe i wykonywane u wszystkich noworodków w pierwszych dniach życia. Polega ono na pobraniu próbki krwi z pięty dziecka i oznaczeniu poziomu fenyloalaniny. Podwyższony poziom fenyloalaniny wskazuje na możliwość wystąpienia PKU i wymaga dalszej diagnostyki.
Wczesne wykrycie PKU i natychmiastowe wdrożenie leczenia dietetycznego pozwala na uniknięcie poważnych powikłań neurologicznych i zapewnienie dziecku prawidłowego rozwoju. Dieta w PKU polega na ograniczeniu spożycia fenyloalaniny, poprzez eliminację lub znaczne ograniczenie pokarmów bogatych w białko, takich jak mięso, ryby, jaja, nabiał i orzechy. Dieta musi być ściśle kontrolowana przez lekarza i dietetyka, a pacjent musi spożywać specjalne preparaty aminokwasowe, które dostarczają organizmowi niezbędne aminokwasy bez fenyloalaniny.
Przestrzeganie diety jest szczególnie ważne w okresie dzieciństwa i dojrzewania, kiedy mózg intensywnie się rozwija. W przeszłości uważano, że po okresie dojrzewania dieta może być mniej restrykcyjna, jednak obecnie zaleca się kontynuowanie diety przez całe życie, aby utrzymać stabilny poziom fenyloalaniny we krwi i zapobiec potencjalnym problemom zdrowotnym w późniejszym wieku, takim jak problemy z koncentracją, pamięcią i nastrojem.
Szczególną uwagę należy zwrócić na kobiety z PKU planujące ciążę. Wysoki poziom fenyloalaniny we krwi matki może być toksyczny dla rozwijającego się płodu i prowadzić do poważnych wad wrodzonych, nawet jeśli dziecko nie odziedziczy PKU. Dlatego kobiety z PKU powinny skonsultować się z lekarzem specjalistą i dietetykiem przed planowaną ciążą i ściśle przestrzegać diety w trakcie ciąży. W niektórych przypadkach konieczne może być wprowadzenie dodatkowych środków ostrożności, takich jak częstsze monitorowanie poziomu fenyloalaniny i dostosowanie diety.
Oprócz diety, w leczeniu PKU stosuje się również leki, które pomagają obniżyć poziom fenyloalaniny we krwi. Jednym z leków stosowanych w PKU jest sapropteryna (tetrahydrobiopteryna, BH4), która jest koenzymem hydroksylazy fenyloalaninowej. Sapropteryna może zwiększyć aktywność resztkowej hydroksylazy fenyloalaninowej u niektórych pacjentów z PKU, co pozwala na zwiększenie tolerancji na fenyloalaninę w diecie. Jednak nie wszyscy pacjenci z PKU reagują na sapropterynę, a skuteczność leku zależy od rodzaju mutacji genu PAH.
Nowe metody leczenia PKU, takie jak terapia genowa i terapia mRNA, są obecnie w fazie badań klinicznych i obiecują potencjalne wyleczenie choroby w przyszłości. Terapia genowa polega na wprowadzeniu prawidłowej kopii genu PAH do komórek pacjenta, co umożliwia produkcję funkcjonalnego enzymu. Terapia mRNA natomiast polega na wprowadzeniu do komórek mRNA, który koduje hydroksylazę fenyloalaninową, co również prowadzi do produkcji funkcjonalnego enzymu.
Podsumowując, fenyloketonuria jest chorobą genetyczną, która wymaga wczesnego wykrycia i leczenia, aby zapobiec poważnym powikłaniom. Dziedziczenie autosomalne recesywne oznacza, że oboje rodzice muszą być nosicielami zmutowanego genu, aby dziecko urodziło się z PKU. Badania przesiewowe noworodków, dieta i leki są skutecznymi metodami leczenia PKU i pozwalają na zapewnienie pacjentom prawidłowego rozwoju i jakości życia. Rozwój nowych terapii, takich jak terapia genowa i terapia mRNA, daje nadzieję na potencjalne wyleczenie choroby w przyszłości. Wiedza na temat dziedziczenia, diagnostyki i leczenia PKU jest kluczowa dla lekarzy, pacjentów i ich rodzin, aby zapewnić jak najlepszą opiekę i wsparcie.